R包安装为何总报错
时间:2017-08-31 来源:原创 作者:森莘
微信公众号:biowolf_cn 点击:次
| 经常由学员反应,R包安装出错,问题可能由很多种,我们先看下正确的安装方法。 一、安装R包方法 1、首先我们来认识一下bioconductor的命令,这是下载所有bioconductor R包需要先输入的一条命令:source("http://bioconductor.org/biocLite.R") 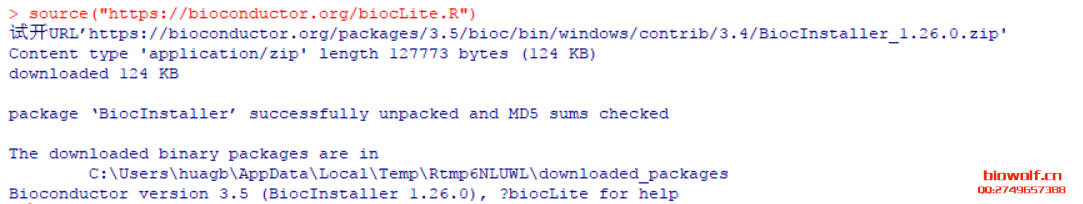 2、试着安装一个edgeR包  大家应该注意到,上面老师给大家标注的地方,我们R安装R包,首先回下载R包,这里需要检查下载的R包是否和需要下载的大小相同,其实这些语句都很简单,但很多学员看都不看,报错就截图给老师,说怎么安装不了,其实可能是你的网速不好,根本没有完整的下载R包,这一点非常重要。 在安装R包时经常出现:Updata all/some/none?[a/s/n]: 遇到这句命令很多学员就不知道如何操作了,这句命令的字面意思也很简单,就是问软件的使用者,是否需要更新其他R包,这里一律输入“n”,当然如果你的时间够充裕,也可以输入a全部更新试试,不过这个不是必要的,下次记得了,一律输入n就搞定了。 R这个软件挺智能,会自动安装相关的R包,看到上面的截图,我们可以看到安装edgeR包时也安装了limma、locfit包,还有些包需要安装十几个相关联的R包,这个时候就要耐心等待,不要心急,有些学员R还在下载,就截图说怎么还不行,这个要等R安装好所有的关联包。 3、检查R包是否安装成功:library("edgeR") ,你也可以输入你需要检验的R包,发现没有报错,说明R包安装成功。 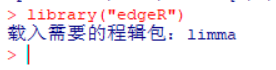 4、大众R包的安装:install.packages("gplots") 这里由学员就由疑问,如何区分大众R包和bioconductor R包呢,其实这个不需要记住的,如果你使用的别人写的R包,那么代码会标注相关R包的下载安装;如果时自己写的代码,那么你肯定知道该包哪个类型,因为你在写程序时,已经对这个R包做过很多分析,否则你也不知道如何使用。 需要记住的就是这两种类型的包,在安装时命令时不同的,大部分都是大众包,生物信息分析的专业包,才是由bioconductor发布的。 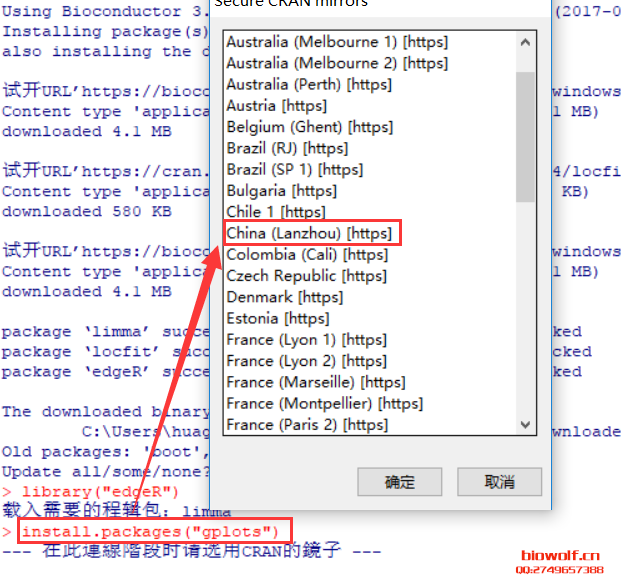 安装大众包,有个选择镜像的过程,一般情况下,我们选择中国离自己省份近的镜像,当然选择第一个,就是总服务器的也是可以的,前提时网速够快。 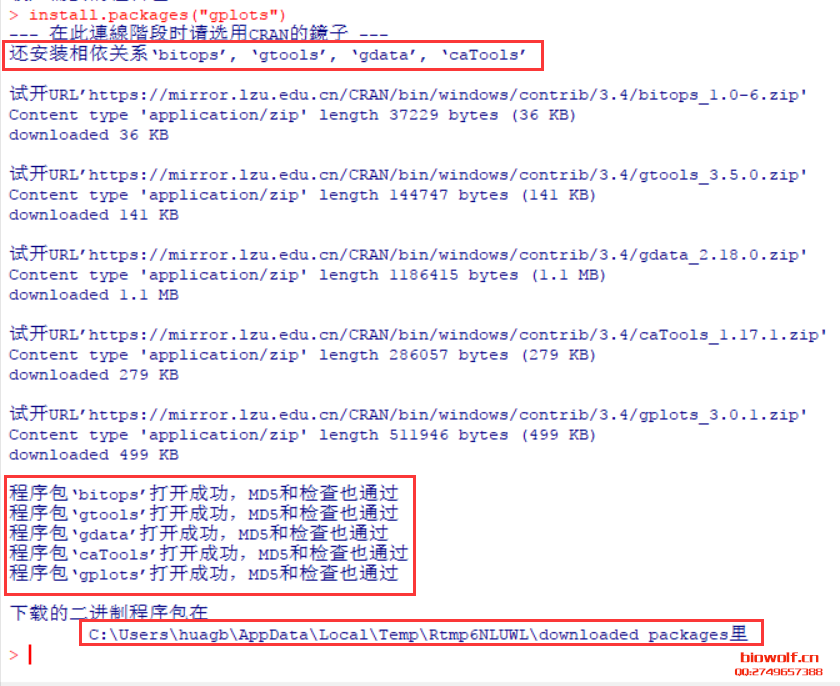 调用R包  二、常见问题 1、电脑配置 如果电脑配置太低,请勿安装最新版本,在选择R版本时,需要根据操作系统选择,常用的win系统就选择win系统的R。 如果担心出错,安装比最新版第一个版本的R,因为当R出新版时,很多R包是没有更新的,这样就可能会报错,比如现在组新版是R3.4.1,那么我们安装R 3.4.0就是一个不错的选择。 2、工作目录的设置 如果不想麻烦,直接安装在C盘,很多学员操作习惯很好,把软件都安装在非系统盘,这样有可能报错。这里需要对安装好的工作目录做一个设置,这个步骤可以减少很多错误,因为我们R下载的R包,需要解压到R工作目录的“library”这个文件夹内,如果电脑不允许在这个文件夹读写,那么肯定要报错; 解决方法:设置工作目录的权限,把所有用户都设置为完全控制。 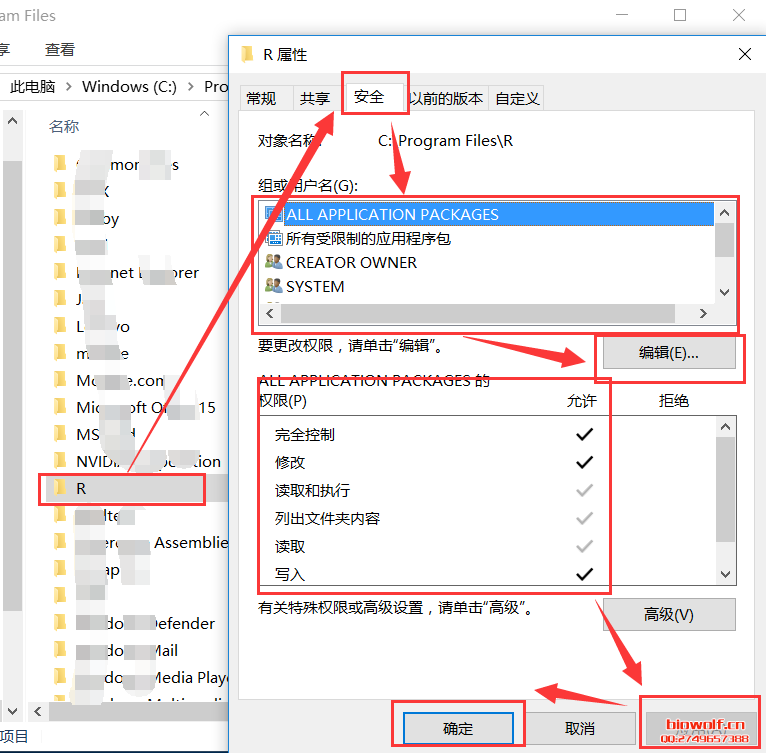 3、这个比较少见,就是R所有的R包都是使用IE浏览器下载的,而极少学员的IE浏览器无法打开R包的链接,所以安装就报错,这个时候需要设置代理路径。 (责任编辑:伏泽 微信:18520221056) 
|
- 上一篇:如何获取生信分析课程代码?
- 下一篇:学习TCGA数据库挖掘基础课程常见问题