|
下载本文文献,关注微信公众号:biowolf_cn,回复“免疫浸润” 1. 去卷积方法 CIBERSORT  网址:https://cibersort.stanford.edu/ CIBERSORT是2015年在Nature Methods发表的一个方法,现在被引量为84,其方法摘要为: a method for characterizing cell composition of complex tissues from their gene expression profiles. When applied to enumeration of hematopoietic subsets in RNA mixtures from fresh, frozen, and fixed tissues, including solid tumors, CIBERSORT outperformed other methods with respect to noise, unknown mixture content, and closely related cell types. CIBERSORT should enable large-scale analysis of RNA mixtures for cellular biomarkers and therapeutic targets (http://cibersort.stanford.edu). 在Cell Reports那篇文章提供的TCIA网址中得到了关于LM22的细胞占比数据,之前不理解,这篇文章有详细的说明: To assess the feasibility of leukocyte deconvolution from bulk tumors, we designed and validated a leukocyte gene signature matrix, termed LM22. It contains 547 genes that distinguish 22 human hematopoietic cell phenotypes, including seven T cell types, naïve and memory B cells, plasma cells, NK cells, and myeloid subsets (Supplementary Table 1, Supplementary Fig. 2, and Online Methods). Cell subsets can be further grouped into 11 major leukocyte types based on shared lineage (Supplementary Table 1). 虽然在这篇文章末尾提到了 We anticipate that CIBERSORT will prove valuable for analysis of cellular heterogeneity in microarray or RNA-Seq data derived from fresh, frozen, and fixed specimens, thereby complementing methods that require living cells as input. 表明文章发表时,CIBERSORT还没有用于RNAseq数据(这点在TIMER中以一个优势被提及)。但从Cell Reports文章《Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade》创建的TCIA网站来看,CIBERSORT方法已经应用到了TCGA的RNAseq数据上。 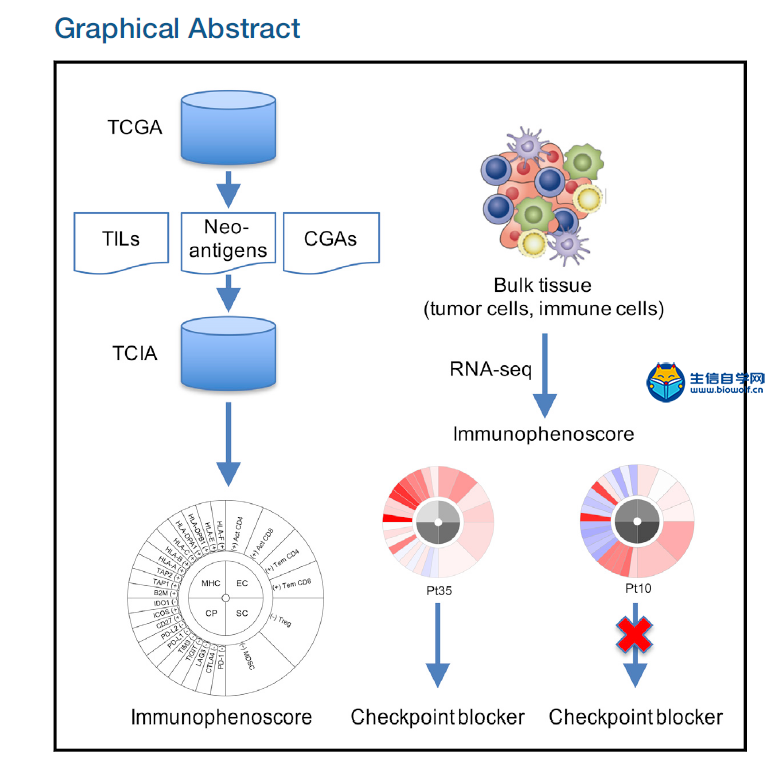 TIMER  《Comprehensive analyses of tumor immunity: implications for cancer immunotherapy》是2016年发表于Genome Biology的一篇计算免疫浸润的方法。 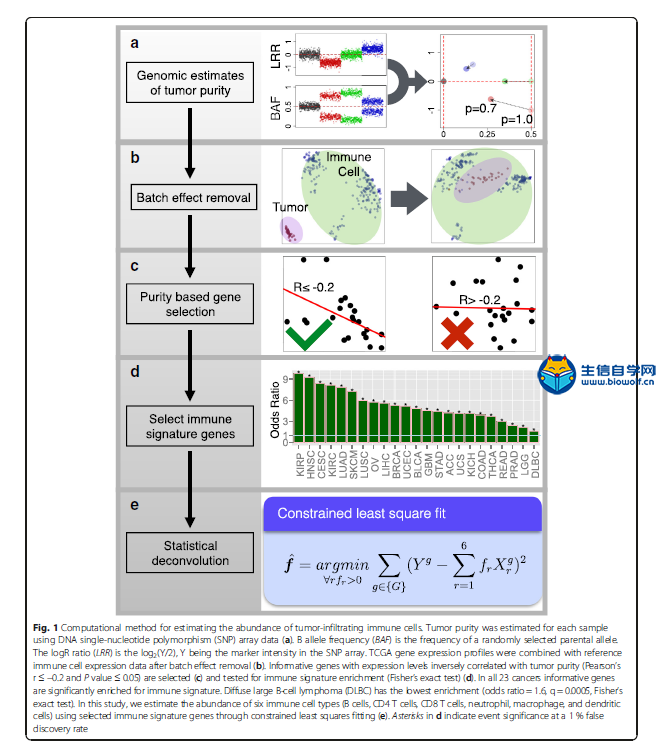 它的主要工作结果为: We developed a computational method to estimate the abundance of six tumor-infiltrating immune cell types (B cells, CD4 T cells, CD8 T cells, neutrophils, macrophages, and dendritic cells) to study 23 cancer types in The Cancer Genome Atlas (TCGA) 设计的大致流程如下: The prerequisite data include tumor purity, tumor gene expression profiles (as transcript per million reads (TPM)) from TCGA, and an external reference dataset of purified immune cells. Tumor purity is critical to selecting genes informative for deconvolving immune cells in the tumor tissue and was inferred from copy number alteration data using an R package, CHAT, we have developed [15] (Fig. 1a). Our purity estimation method has been validated using diluted series with known tumor/normal mixture ratios and agreed with previous methods and orthogonal estimations [16]. The distributions of tumor purity showed large variations among different samples across most of the 23 TCGA cancer types (Additional file 1: Figure S1). For each cancer dataset, batch effects between TCGA and the external reference data sets were removed using ComBat [17] (Fig. 1b). Next we selected genes whose expression levels are negatively correlated with tumor purity (Fig. 1c; Additional file 1: Figure S2; Additional file 2: Table S1), an indication that these genes are expressed by stromal cells in the tumor microenvironment. Across all 23 cancers, informative genes selected from the above steps are significantly enriched for a predefined immune signature [18] (Fig. 1d). This result indicates that large numbers of immune cell-specific genes are highly expressed in the tumor microenvironment. Finally, we used constrained least squares fitting [19] on the informative immune signature genes to infer the abundance of the six immune cell types (Fig. 1e). 它指出了自己开发的方法相对于CIBERSORT的优点: Our work first provided a systematic prognostic landscape of different tumor-infiltrating immune cells in diverse cancer types. We compared our results with two recent studies on the same topic [6, 11]. The method used in Gentles et al., CIBERSORT [35], is currently only applicable to microarray data, thus unable to analyze the TCGA RNA-seq data. Therefore, our immune component estimation is a unique addition to TCGA for future integrative analyses of tumor–immune interactions. By including more immune cell types into regression, CIBERSORT inference also suffered from statistical co-linearity that might have resulted in biased estimations (Additional file 7: Table S6; Additional file 8). Due to this limitation, although Gentles et al. studied more cell types, they reported few significant prognostic immune predictors, without correction for other clinical confounders. In contrast, we observed many more significant clinical associations with the correction of multiple cofactors. 值得注意的是,在文末提供的方法部分,作者详细地介绍了方法的计算流程,重点强调了 To note, coefficients f estimated using this method are the relative abundance of immune cells. The scale of the estimation of an individual immune cell type is determined by the variance of the corresponding reference data Xr. Therefore, f are not comparable between cancer types or different immune cells. Source codes for TIMER and downstream statistical analysis as well as related data files are available at http://cistrome.org/TIMER/download.html. 说明最后的计算结果是一个系数,它表明的只是免疫细胞相对的丰富度。 2. GSEA 个人认为最值得关注的还是Cell Reports的文章Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade。它于2017年发表,本身的背景中就有提及前面两种方法去卷积方法。 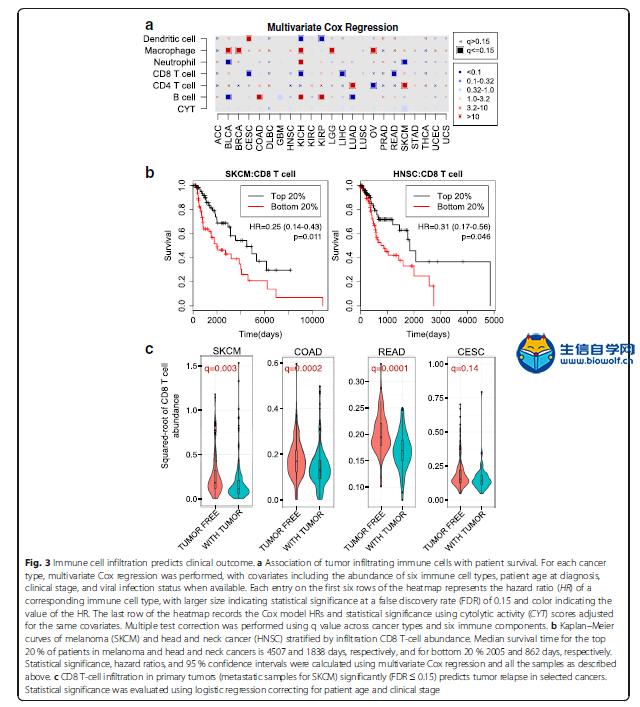 文章对TIMER的观点是: RNA expression data corrected for tumor purity were used to estimate infiltration of B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells ([Li et al., 2016](javascript:void(0);)). However, although such analyses of few major cell types are helpful for identifying clinical associations, higher resolution of the TIL landscape is required in order to dissect tumor-immune cell interactions and identify prognostic and predictive markers. 因此,该文章提供了更为细致的免疫浸润组分图景,有近30种细胞类型 Thus, it is of utmost importance to provide a comprehensive view of the intratumoral immune landscape including memory cells, cytotoxic cells (CD8+ T cells, natural killer [NK] cells, and NK T [NKT] cells), as well as immunosuppressive cells (Tregs and myeloid-derived suppressor cells [MDSCs]). we estimated 28 subpopulations of TILs including major types related to adaptive immunity: activated T cells, central memory (Tcm), effector memory (Tem) CD4+ and CD8+ T cells, gamma delta T (Tγδ) cells, T helper 1 (Th1) cells, Th2 cells, Th17 cells, regulatory T cells (Treg), follicular helper T cells (Tfh), activated, immature, and memory B cells, as well as cell types related to innate immunity, such as macrophages, monocytes, mast cells, eosinophils, neutrophils, activated, plasmacytoid, and immature dendritic cells (DCs), NK cells, natural killer T (NKT) cells, and MDSCs. 在构建的TCIA中,它使用了CIBERSORT去卷积方法进行免疫浸润的计算,方法是通过将TCGA的RNAseq数据转化为CIBERSORT能够处理的MicroArray数据集。 重点是,该文章提供了一种新的免疫浸润细胞组分计算的思路:就是使用基因富集分析。 下面介绍了该方法及其优点 Our approach is based on the use of metagenes, i.e., non-overlapping sets of genes that are representative for specific immune cell subpopulations and are neither expressed in CRC cell lines nor in normal tissue. The expression of these sets of metagenes is then used to analyze statistical enrichment using gene set enrichment analysis (GSEA). The advantage of the metagene approach is the robustness of the method due to two characteristics: (1) the use of a set of genes instead of single genes that represent one immune subpopulation, because the use of single genes as markers for immune subpopulations can be misleading as many genes are expressed in different cell types; and (2) the assessment of relative expression changes of a set of genes in relation to the expression of all other genes in a sample. Thus, the calculations are less sensitive to noise resulting from sample impurity or sample preparation compared with the deconvolution methods. 这两种方法都能够在TCIA上使用: We built a model to transform RNA-sequencing data to microarray data considering the TCGA samples for which microarray and RNA-sequencing data were available (n = 550; see Experimental Procedures). We present here only the results from the GSEA method, which depicts a more comprehensive picture of the tumor-suppressive or tumor-promoting roles of TILs. However, we make both GSEA and deconvolution data available on the TCIA website.  Enrichment bubble plot.png 该方法提供了免疫细胞在病人样本中的富集度(上图是以绝对数目显示,也可以显示富集的相对比例),通过NES(averaged normalized enrichment score)与FDR进行样本的预筛。默认设定为NES>0, Q-value(FDR) < 0.1。可以下载相应的文件,包含样本名(Barcode),NES和Q-value 3列。 入门免疫浸润,可以学习生信自学网推出的免疫浸润系列课程: TCGA肿瘤免疫细胞浸润模式挖掘 GEO数据库免疫细胞浸润视频 甲基化免疫细胞浸润模式 TCGA数据库肿瘤微环境 TCGA数据库肿瘤突变负荷 下载本文文献,关注微信公众号:biowolf_cn,回复“免疫浸润” 
责任编辑:伏泽 作者申明:本文版权属于生信自学网(微信号:18520221056)未经授权,一律禁止转载! |




