|
肿瘤免疫一下子成了2019年的分析热点,下面生信自学网给大家介绍一篇高分肿瘤微环境的文献: Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures 胃癌肿瘤微环境特征鉴定预后和免疫治疗相关基因特征 如果需要做生信定制分析报告,希望参考这篇文献,结合自己的研究方向做肿瘤微环境分析,可以联系微信:18520221056 文章后面还提供文献下载链接哦 GC: Gastric cancer 胃癌 TME: Tumor microenvironment 肿瘤微环境 PCA: Principal component analysis 主成分分析 STAD: Stomach adenocarcinoma 胃腺癌 TPM: Transcripts Per Kilobase Million 每千碱基百万的转录本 FPKM: Fragments per kilobase million 每千克碎片百万 CIBERSORT: Cell type identification by estimating relative subset of known RNA transcripts 通过估计已知RNA转录物的相对子集来识别细胞类型 TMEscore: TME signature score 肿瘤微环境打分 GO: Gene ontology 基因本体论 GSEA: Gene set enrichment analysis 基因集富集分析 KEGG: Kyoto encyclopedia of genes and genomes 京都基因和基因组百科全书 FDR: False discovery rate 错误发现率 CYT: Cytolytic activity 细胞溶解活性 DEG: Differentially expressed gene 差异表达基因 MCP-counter: Microenvironment Cell Populations-counter 微环境细胞数量计数器 MSI: Microsatellite instability 微卫星不稳定性 EBV: Epstein-Barr virus 爱泼斯坦巴尔病毒 EMT: Epithelial-mesenchymal-transition 上皮间充质转变 GS: Genomically stable 基因稳定 CIN: Chromosomal instability 染色体不稳定 CR: Complete response 完全响应 PR: Partial response 部分响应 SD: Stable disease 稳定疾病 PD: Progressive disease 进行性疾病 TMB: Tumor mutational burden 肿瘤突变负荷 如果需要学习TCGA肿瘤免疫细胞浸润模式挖掘,可以购买课程《TCGA肿瘤免疫浸润》 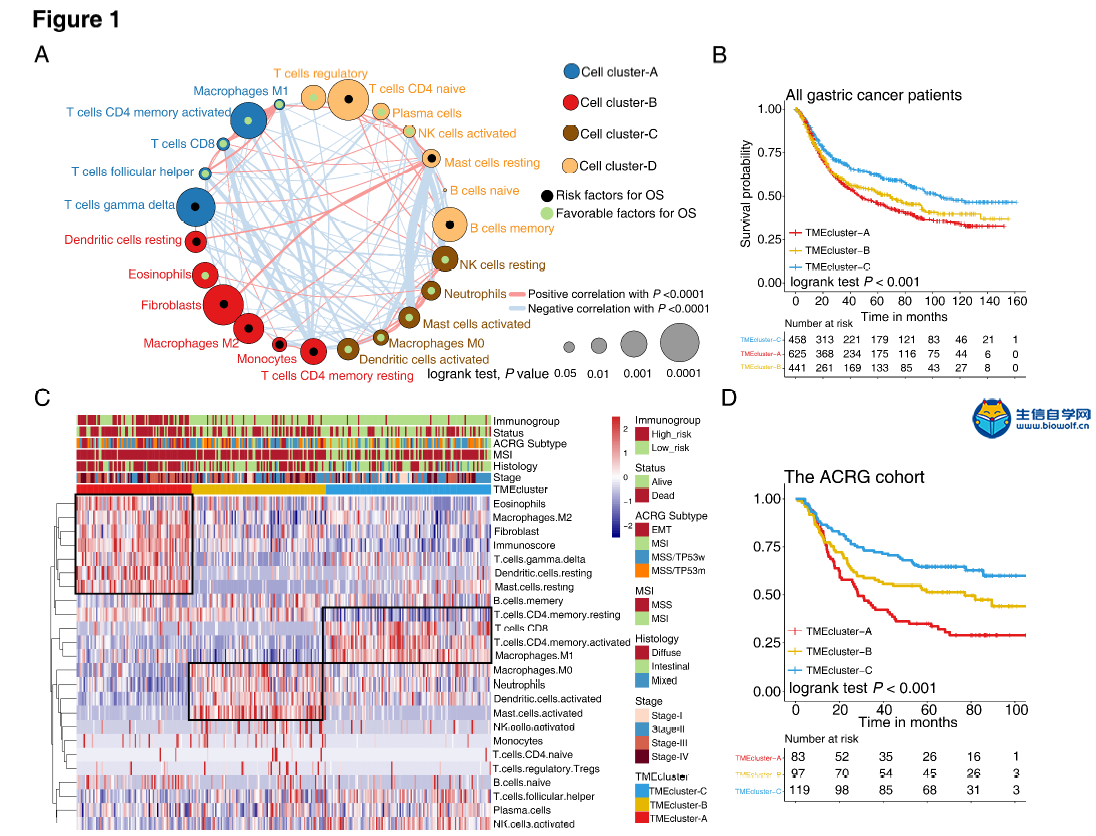 ABSTRACT Tumor microenvironment (TME) cells constitute a vital element of tumor tissue. Increasing evidence has elucidated their clinicopathological significance in predicting outcomes and therapeutic efficacy. Nonetheless, no studies have reported a systematic analysis of cellular interactions in the tumor microenvironment. In this study, we comprehensively estimated the TME infiltration patterns of 1,524 gastric cancer patients and systematically correlated the TME phenotypes with genomic characteristics and clinicopathological features of gastric cancer using two proposed computational algorithms. Three TME phenotypes were defined, and the TMEscore was constructed using principal component analysis algorithms. The high TMEscore subtype was characterized by immune activation and response to virus and interferon-gamma. Additionally, activation of transforming growth factor β, epithelial mesenchymal transition, and angiogenesis pathways were observed in the low TMEscore subtype, which are considered T-cell suppressive and may be responsible for significantly worse prognosis in gastric cancer (hazard ratio [HR], 0.42; 95% confidence interval [CI], 0.33–0.54; P < 0.001). Furthermore, multivariate analysis revealed that TMEscore was an independent prognostic biomarker, and its value in predicting immunotherapeutic outcomes was also confirmed (IMvigor210 cohort: HR, 0.63; 95% CI, 0.46–0.89; P = 0.008; GSE78220 cohort: HR, 0.25; 95% CI, 0.07–0.89; P = 0.021). Depicting a comprehensive landscape of the TME characteristics of gastric cancer may therefore help to interpret the responses of gastric tumors to immunotherapies and provide new strategies for the treatment of cancers. 摘要 肿瘤微环境(TME)细胞是肿瘤组织的重要组成部分。越来越多的证据阐明了它们在预测预后和疗效方面的临床病理学意义。尽管如此,还没有研究报告对肿瘤微环境中的细胞相互作用进行了系统的分析。在本研究中,我们综合评估了1524例胃癌患者的TME浸润模式,并使用两种建议的计算算法,系统地将TME表型与胃癌的基因组特征和临床病理特征相关联。定义了三个TME表型,并利用主成分分析算法构建了TME谱。高TME score亚型的特点是免疫激活和对病毒和干扰素γ的反应。此外,在低TME score亚型中观察到转化生长因子β的激活、上皮间质转化和血管生成途径,这些亚型被认为是T细胞抑制,可能导致胃癌预后显著恶化(危险比[HR],0.42;95%置信区间[CI],0.33–0.54;P< 0.001)。此外,多变量分析显示,TME score是一个独立的预后生物标志物,其在预测免疫治疗结果方面的价值也得到了证实(IMvigor 210队列:HR,0.63;95%CI,0.46–0.89;P=0.008;GSE78220队列:HR,0.25;95%CI,0.07–0.89;P=0.021)。因此,全面描述胃癌的TME特征有助于解释胃癌对免疫治疗的反应,并为癌症的治疗提供新的策略。 需要学习TCGA肿瘤微环境分析,可以购买生信自学网课程《TCGA数据库肿瘤微环境》 Introduction Genomic analysis has been the primary methodology used in international efforts to discover novel biological targets in gastric cancer (1,2), although this method has not led to the successful discovery of distinct mechanisms. However, some studies have revealed the significance of tumor-related structures as well as upregulated signaling pathways in both cancer cells and the tumor microenvironment (TME) (3,4), suggesting that intercellular relationships are more important than genomic factors at the single-cell level (5,6). In addition, an increasing body of literature suggests a crucial role for the TME in cancer progression and therapeutic responses (7,8). For example, differences in the compositions of resident cell types within the TME, including cytotoxic T cells, helper T cells, dendritic cells (DCs), tumor-associated macrophages (TAMs), mesenchymal stem cells (MSCs), and associated inflammatory pathways have been reported in patients with cancer (5,6,9,10). The TME context determined at diagnosis reflects the immune response (11) and chemotherapy benefit (8), and changes in the numbers of CD8+ T cells, CD4+ T cells, macrophages, and cancer-associated fibroblasts infiltrating in the TME correlate with clinical outcomes in various malignancies, including gastric cancer, melanoma, urothelial cancer, lung cancer, and breast cancer (10,12-14). Because gastric cancers are significantly associated with infectious agents, most notably Helicobacter pylori and Epstein-Barr virus (EBV), biomarkers that can predict responsiveness to immune-checkpoint blockade are being extensively investigated to further improve precision immunotherapy (15). Moreover, the abundance of immune cells and other cells in the TME can be estimated using computational methods (16-18). Although several studies using these methodologies have explored the clinical utility of TME infiltrates (7,19), and although several mechanisms associated with the role of TME in immunotherapy response and resistance have been experimentally identified for some tumor types (4,13,14,20,21), to date, the comprehensive landscape of cells infiltrating the TME has not yet been elucidated. In the present study, two proposed computational algorithms (16,17) were employed to estimate the fractions of 22 immune cell types and cancer-associated fibroblasts based on clinically annotated gastric cancer gene expression profiles (1,22). We estimated the TME infiltration patterns of 1524 tumors from patients with gastric cancer and systematically correlated the TME phenotypes with genomic characteristics and clinical and pathological features of gastric cancer. As a result, we established a methodology to quantify the TME infiltration pattern (TMEscore). TMEscore was found to be a robust prognostic biomarker and predictive factor for response to immune-checkpoint inhibitors. 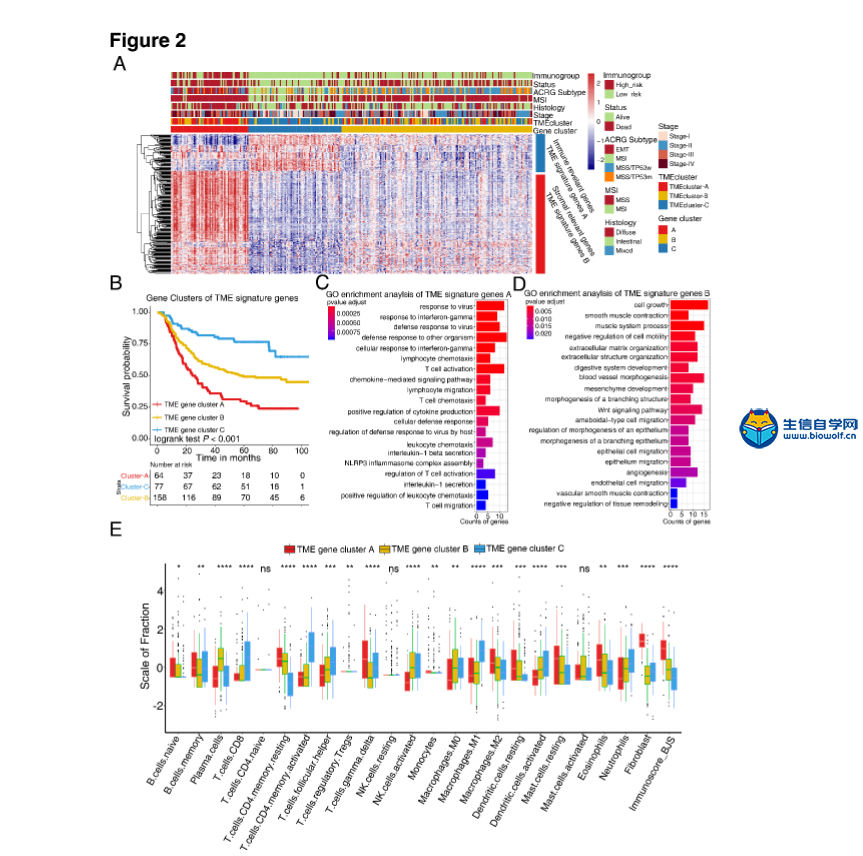 介绍 基因组分析已成为国际上探索胃癌新生物学靶点(1,2)的主要方法,尽管这种方法尚未成功发现不同的机制。然而,一些研究揭示了肿瘤相关结构的重要性以及在癌细胞和肿瘤微环境(TME)(3,4)中上调的信号通路,表明细胞间关系比单细胞水平的基因组因子更重要(5,6)。此外,越来越多的文献表明,TME在癌症进展和治疗反应中起着至关重要的作用(7,8)。例如,在癌症患者(5,6,9,10)中,报告了TME内常驻细胞类型的组成的差异,包括细胞毒性T细胞、辅助T细胞、树突状细胞(DCs)、肿瘤相关巨噬细胞(TAMs)、间充质干细胞(MSCs)和相关炎症途径。诊断时确定的TME背景反映了免疫反应(11)和化疗益处(8),TME中CD8+T细胞、CD4+T细胞、巨噬细胞和肿瘤相关成纤维细胞浸润数量的变化与各种恶性肿瘤的临床结果相关,包括胃癌、黑色素瘤、尿路上皮癌、肺癌和乳腺癌。 由于胃癌与感染因子,尤其是幽门螺杆菌和爱泼斯坦-巴尔病毒(EBV)显著相关,可以预测免疫检查点阻断反应的生物标志物正在被广泛研究,以进一步提高免疫治疗的精确度(15)。此外,TME中免疫细胞和其他细胞的丰度可用计算方法(16-18)估算。尽管使用这些方法进行的几项研究已经探索了TME浸润的临床效用(7,19),尽管迄今为止,一些肿瘤类型(4,13,14,20,21)已经通过实验确定了与TME在免疫治疗反应和抵抗中的作用相关的几种机制,渗透TME的细胞的综合前景尚未阐明。 在本研究中,基于临床注释的胃癌基因表达谱(1,22),采用两种建议的计算算法(16,17)来估计22种免疫细胞类型和肿瘤相关成纤维细胞的部分。我们估计了1524例胃癌患者的TME浸润模式,并系统地将TME表型与胃癌的基因组特征、临床和病理特征联系起来。因此,我们建立了一个量化TME渗透模式(TMESCORE)的方法。TMESCORE被发现是一个强有力的预后生物标志物和免疫检测点抑制剂反应的预测因子。 Materials and Methods Gastric cancer datasets and preprocessing We systematically searched for gastric cancer gene expression datasets that were publicly available and reported full clinical annotations. Patients without survival information were removed from further evaluation. In total, we gathered six treatment-naive cohorts of samples from patients with gastric cancer for this study: ACRG/GSE62254, GSE57303, GSE84437, GSE15459, GSE26253, GSE29272, and TCGA-STAD. The raw data from the microarray datasets generated by Affymetrix and Illumina were downloaded from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). The raw data for the dataset from Affymetrix were processed using the RMA algorithm for background adjustment in the Affy software package (23). RMA was used to perform background adjustment, quantile normalization, and final summarization of oligonucleotides per transcript using the median polish algorithm. The raw data from Illumina were processed using the lumi software package. Data from The Cancer Genome Atlas (TCGA) were downloaded from the UCSC Xena browser (GDC hub), as detailed in the supplementary methods. For TCGA dataset, RNA-sequencing data (FPKM values) were transformed into transcripts per kilobase million (TPM) values, which are more similar to those resulting from microarrays and more comparable between samples (24). The criteria used for dataset selection, platform and source of each dataset, numbers of samples, and clinical end points are summarized in the Supplementary Methods and Supplementary Table S1. Data were analyzed with the R (version 3.4.0) and R Bioconductor packages.  材料和方法 胃癌数据集和预处理 我们系统地搜索了胃癌基因表达数据集,这些数据集是公开的,并报告了完整的临床注释。无生存信息的患者从进一步评估中剔除。本研究共收集了6组胃癌患者的治疗样本:ACRG/GSE62254, GSE57303, GSE84437, GSE15459, GSE26253, GSE29272, and TCGA-STAD。Affymetrix和Illumina生成的微阵列数据集的原始数据从Gene Expression Omnibus(https://www.ncbi.nlm.nih.gov/geo/)下载。在Affy软件包(23)中,使用用于背景调整的RMA算法处理来自Affymetrix的数据集的原始数据。使用RMA进行背景调整、分位数归一化以及使用中位数波兰算法对每个转录物的寡核苷酸进行最终总结。使用lumi软件包处理来自Illumina的原始数据。癌症基因组图集(TCGA)的数据从UCSC Xena浏览器(GDC hub)下载,详情见补充方法。对于TCGA数据集,RNA测序数据(FPKM值)被转换成每千碱基百万(TPM)值的转录物,这与微阵列产生的结果更相似,并且在样本之间更具可比性(24)。数据集选择的标准、每个数据集的平台和来源、样本数量和临床终点总结在补充方法和补充表S1中。使用R(3.4.0版)和R生物导体包分析数据。 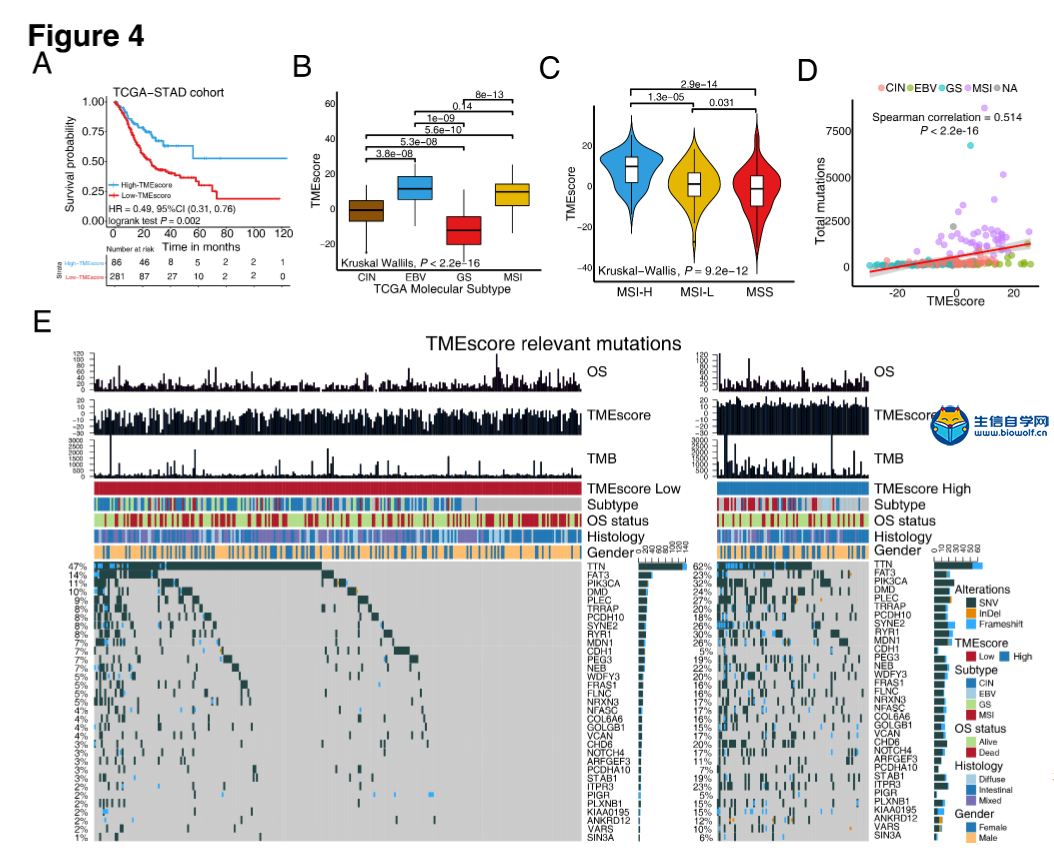 Collection of clinical data The corresponding clinical data from these datasets were retrieved and manually organized when available. For some series, clinical data not attached to gene expression profiles were obtained through one of the following three methods: i) directly downloaded from the corresponding item page in the GEO dataset website, ii) from the supplementary materials in the relative literature, and iii) using the GEOquery package in R. Corresponding authors were contacted for further information where necessary. Updated clinical data and sample information for TCGA-STAD samples were obtained from the Genomic Data Commons (https://portal.gdc.cancer.gov/) using the R package TCGAbiolinks (25). Overall survival information of all TCGA datasets was obtained from the supplementary data of recently published research (26). 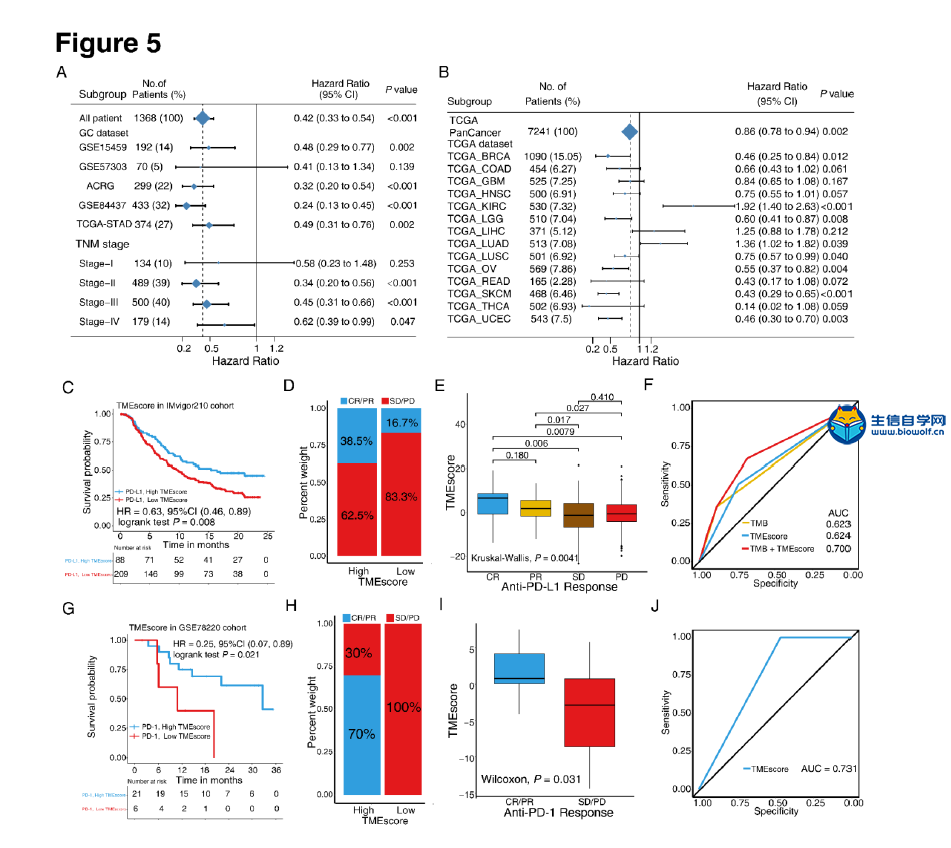 临床资料收集 从这些数据集中检索相应的临床数据,并在可用时手动组织。对于一些系列,未附在基因表达谱上的临床数据是通过以下三种方法之一获得的:i)直接从GEO数据集网站的相应项目页面下载,i i)从相关文献的补充材料下载,以及i i i)使用R中的GEOquery包。必要时联系了相应的作者以获取进一步的信息。更新的TCGA-STAD样本的临床数据和样本信息来自基因组数据共享(https://portal.gdc.cancer.gov/)使用R包TCGAbiolinks(25)。所有TCGA数据集的总体存活信息均来自最近发表的研究补充数据(26)。 文献原文下载:
提取码:3wvb

责任编辑:伏泽 作者申明:本文版权属于生信自学网(微信号:18520221056)未经授权,一律禁止转载! |




