细胞浸润不同工具包的比较仅在过去的两年时间,基于bulk RNA数据反卷积进行不同细胞/组织比例的预测软件开发就很多,我们在网站上一搜索就是一大片文章,其中很多的都是方法学开发的,作为方法学开发的文章,水平都还很高,有NC的,NBT的,各位学员有兴趣的可以自行搜索。
今天分享的文章不是方法学开发的,而是方法学评估的一个文章,文章水平也不低。大家都知道肿瘤微环境中免疫细胞的组成和密度对肿瘤的进展和抗癌治疗的成功有着重要的影响。然而流式细胞术、免疫组织化学染色或单细胞测序由于成本昂贵,常常是不可用的,因此各类RNA-seq反卷积工具包的被开发出来,希望依靠计算方法从bulk RNA-seq数据来估计免疫细胞的组成。今天,我们先一起学习一下《Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology》,了解一下不同工具包的异同。 不同工具包都带有一套自己的细胞类型标记,表中列出每个工具包自带的reference情况。
不同工具包预测细胞组分的性能
这些方法的开发首先就是用来预测不同细胞比例的,所以作者首先比较了他们的预测性能。作者先利用单细胞数据集模拟创建了100个已知细胞类型比例的bulk RNA-seq样本,然后使用不同工具包对模拟数据进行细胞成分预测。结果显示所有方法对B细胞,肿瘤相关成纤维细胞,内皮细胞和CD8+ T 细胞的预测性能都较高(r>0.71); 除了CBS,大多数方法对巨噬细胞/单核细胞,NK细胞和total CD4+ T细胞的预测性能较高(r>0.68);所有方法在区分调节性和非调节性CD4+ T细胞和树突状细胞方面的表现都不佳 (图1a) 。然后作者使用三个独立的数据集对不同方法的性能进行进一步评估,并使用FACS免疫细胞量化作为金标准,结果显示所有预测结果与金标准的结果高度一致 (图1b-d)。 图1
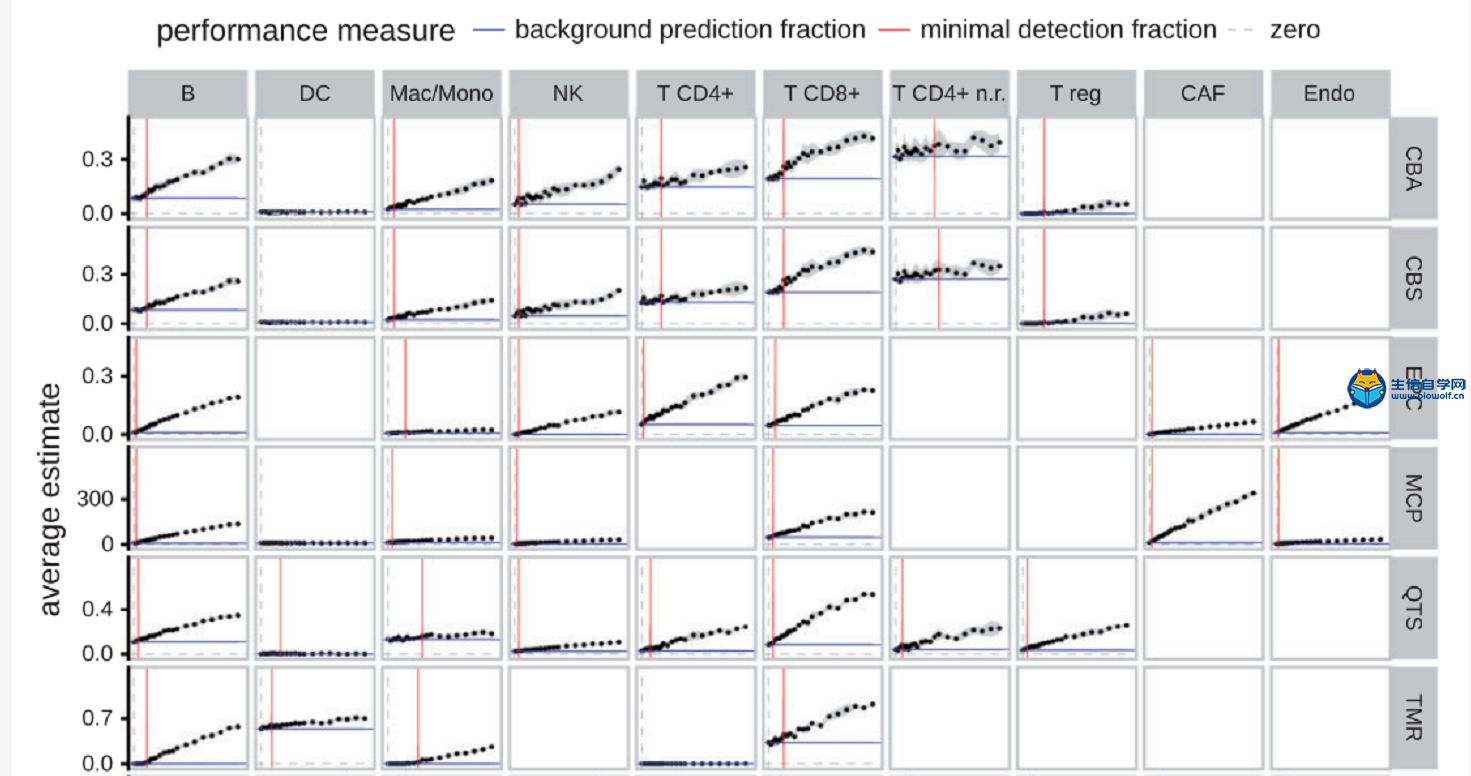
不同工具包的最小检测率和背景预测 为了解决以下两个问题:1)各类工具在免疫细胞何种丰度水平上能够可靠地识别其存在(最小检测率);2)即使一个特定细胞类型实际上不存在,它们可以被预测到多高比例(背景预测)。作者首先从所有其他细胞类型中随机取样的1000个细胞作为背景,并在背景中逐渐添加感兴趣的细胞类型(图2)。
通常情况下,基于反卷积的方法即使在某些细胞不存在的情况下,也会对该类细胞预测一个较低的比例 (图2)。然而EPIC对CAFs,NK细胞,quanTIseq对非调节性CD4+ T细胞和NK细胞的背景预测值很低。TIMER不会受到背景预测的影响(DCs除外),但会以很高的最低检测分数为代价。相比之下,xCell基于marker基因富集的统计方法预测的,它的最小检测分数有略微提高的(大多数细胞类型浸润5%)。简而言之,基于反卷积的方法更容易受到背景预测的影响,这可能是由于某些细胞类型的表达具体相似性(多重共线性),或者是marker基因具有较低的细胞特异性。
图2
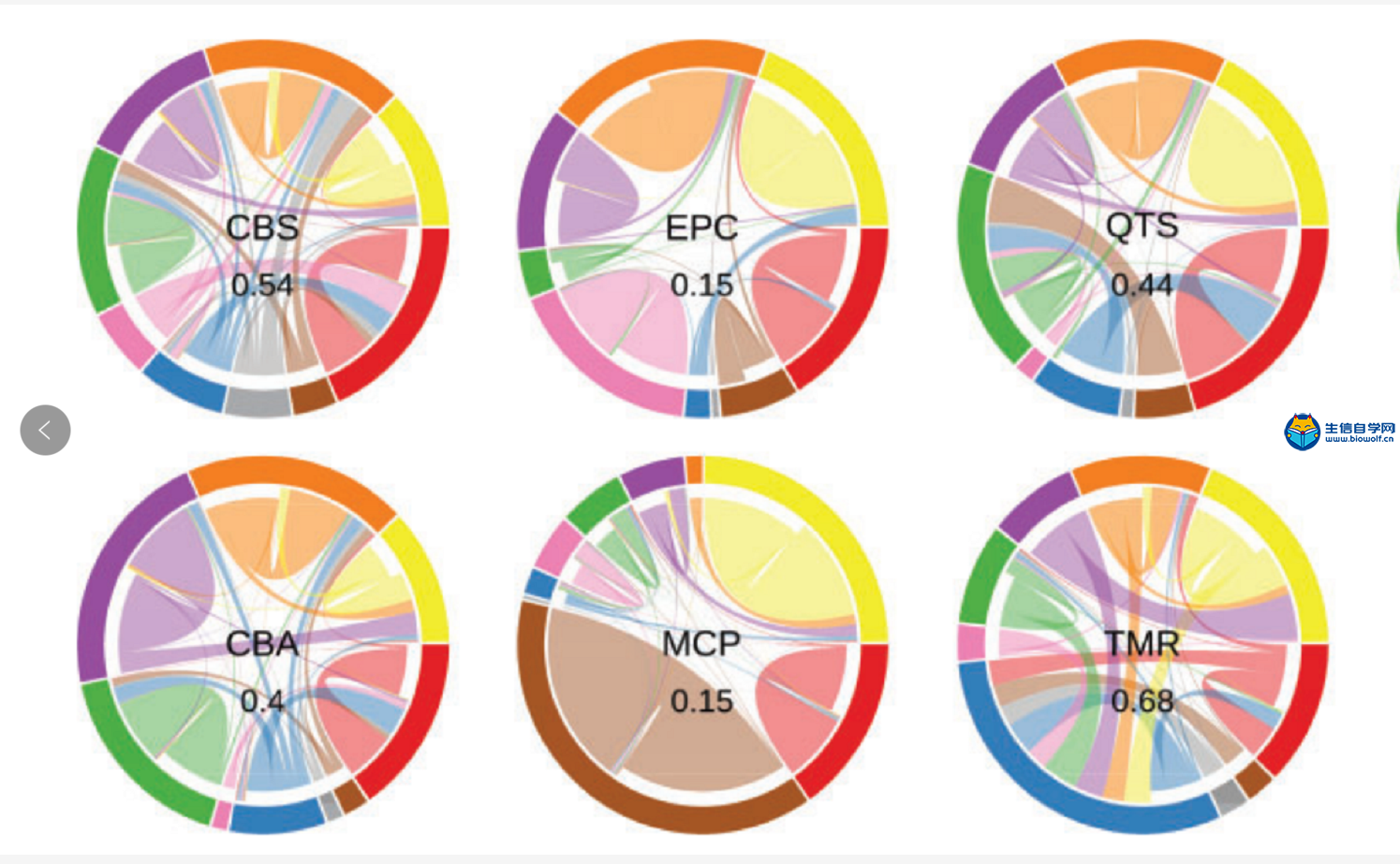
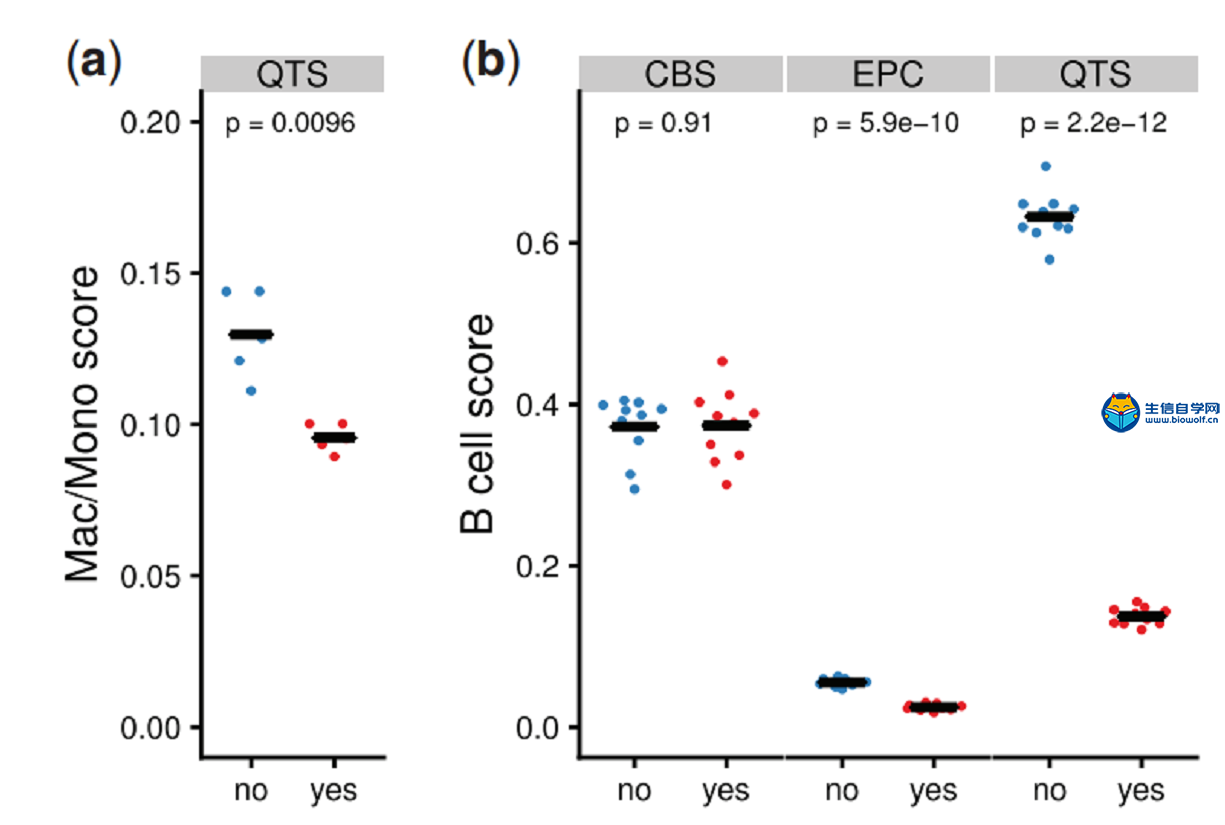
“溢出”分析:背景预测可能是由于非特异marker基因造成的,那么哪种细胞类型会导致工具错误地更高预测另一种细胞的丰度,这种效应称为“溢出”效应。 作者使用含有单一免疫细胞类型的模拟构建bulk RNA-seq样本评估“溢出”效应(图3),并使用经FACS-纯化的免疫细胞进行结果验证。在图2观察到quanTIseq的预测性能受到巨噬细胞/单核细胞高背景预测的影响,图3的“溢出”分析中也注意到显示quanTIseq预测的纯CAF细胞样本中含有大量巨噬细胞/单核细胞。因此,作者怀疑quanTIseq的高背景预测值是由quanTIseq reference矩阵中的非特异性marker基因造成的。事实上作者也鉴定了5个基因,CXCL2, ICAM1, PLTP, SERPING1和CXCL3,它们在CAFs和巨噬细胞/单核细胞中都表达。从reference矩阵中去除这些基因后,背景预测水平显著降低了27%(图4a)。
此外,作者观察到CD8+ T vs CD4+ T、NK vs CD8+ T,DC vs B在所有方法中的“溢出”。对CAF/巨噬细胞进行“溢出“分析,作者确定的六个基因:TCL1A, TCF4, CD37, SPIB, IRF8,BCL11A在B细胞和DC亚群均有表达。事实上,在LifeMap发现数据库中,这6个基因注释为在B细胞和浆细胞样树突状细胞(pDCs)中都表达。当从反褶积矩阵中排除这些基因时,EPIC和quanTIseq的溢出效应显著降低了55%和78%(图4b)。
图3。
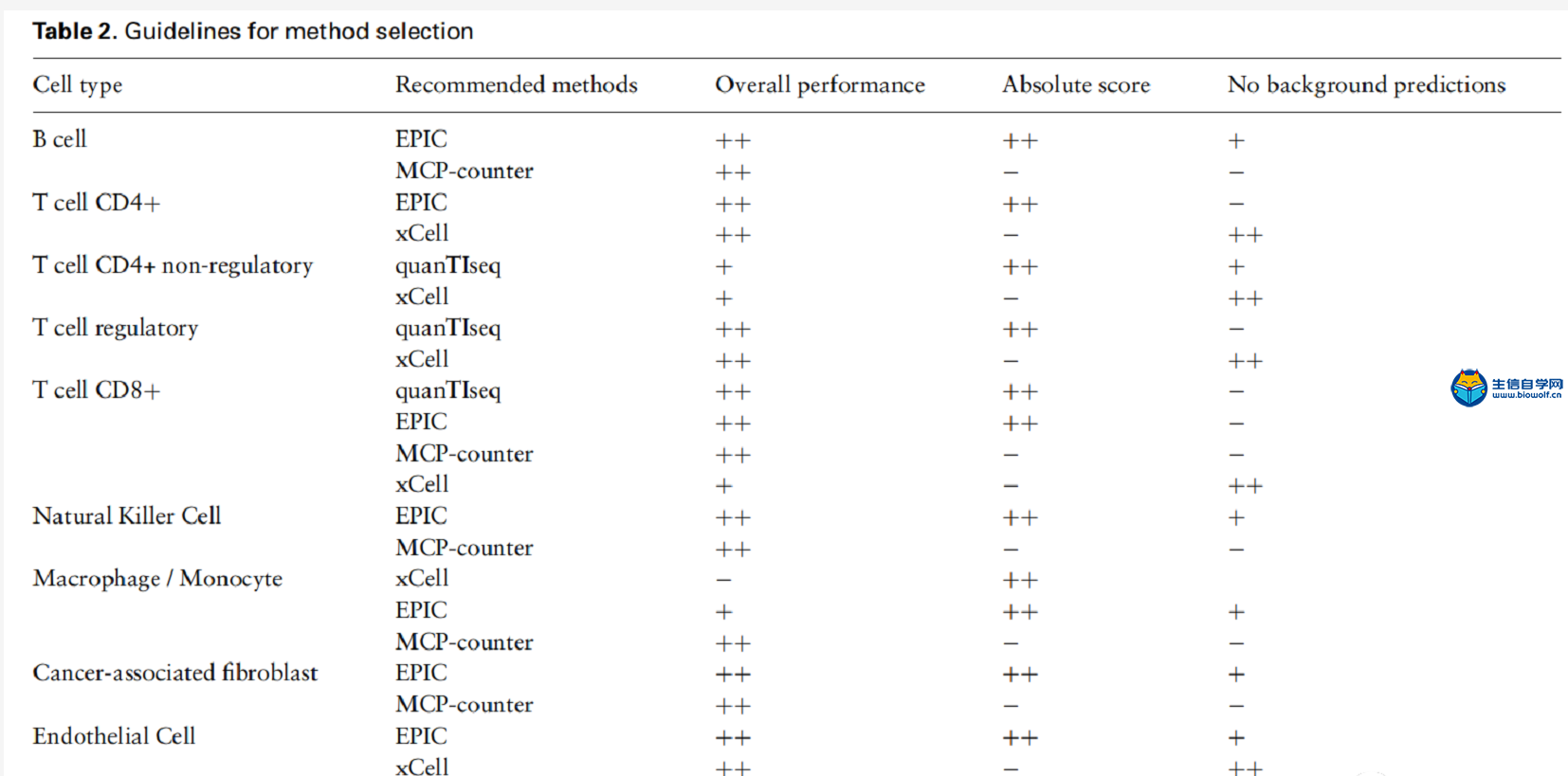
方法选择指南
在表2中,作者根据评分的可解释性、总体性能和可能的局限性三个维度对方法的使用提供指南。EPIC和quanTIseq提供了细胞绝对分数,并且都以任意单位提供细胞组成比列,因此作者推荐使用EPIC和quanTIseq用于一般目的的反褶积。在实践中,绝对细胞比列并不总是必要的,例如,在临床试验中,相对细胞比列也可用于推断治疗组和对照组之间的变化,或用于监测样本纵向免疫成分的变化。这种情况下,使用MCP-counter是一个不错的选择,因为它高度特定的marker在“溢出“分析中表现出色。反褶积方法的一个限制是它们容易受到背景预测的影响,因此,当不存在感兴趣的细胞类型时,作者建议使用xCell。 
责任编辑:伏泽 作者申明:本文版权属于生信自学网(微信号:18520221056)未经授权,一律禁止转载! |