|
肿瘤微环境火了,大家都在想办法把自己的分析向肿瘤微环境靠,然后自己摸索的套路是非常艰辛的,参考已经发表的文章,是生信分析的一个捷径,但是,文献是好,图也很漂亮,然后并没有什么用处,因为很多学员不知道如何做这些分析。零基础学生信就上生信自学网,生信自学网从推出生信视频教程,生信培训,生信答疑以来,一直带领学员披荆斩棘,很多学员也收获满满,不仅论文发表,而且事业小有所成,我们生信自学网在其中也在不断进步,谢谢大家的一路陪伴和支持。 那么正式开始我们今天的主题,首先让我们看看这篇高分生信文章: Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures 胃癌肿瘤微环境特征鉴定预后和免疫治疗相关基因特征 这个是文章的摘要:  然后带大家看看文章基本研究步骤: 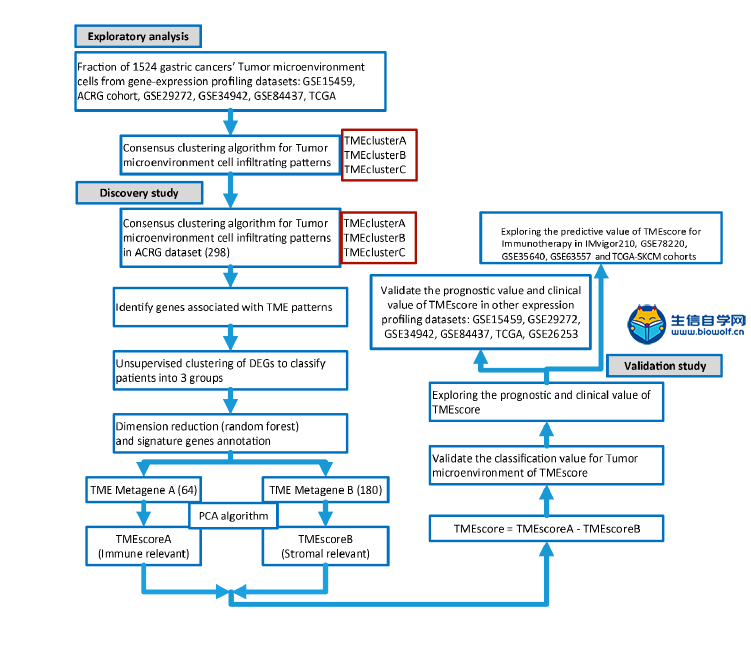 可能大家对文章的分数很好奇,这个大家可以去查一下,一定不会让大家失望的。在这里,如果大家希望结合自己的研究方向,做类似的生信分析,可以联系我们做生信报告合作,合作微信:18520221056 当然大家也可以学习我们推出的肿瘤微环境课程,自己学习入门《TCGA肿瘤微环境》,还可以学习相关课程: 《TCGA肿瘤免疫细胞浸润模式》 《GEO肿瘤免疫细胞浸润》 《TCGA肿瘤突变负荷》 《甲基化免疫细胞浸润基于GEO芯片数据》 不仅可以直接购买课程,还有课程简介和试学课程哦 接下来看看这篇高分文献的研究方法: 材料和方法 胃癌数据集和预处理 我们系统地搜索了胃癌基因表达数据集,这些数据集是公开的,并报告了完整的临床注释。无生存信息的患者从进一步评估中剔除。本研究共收集了6组胃癌患者的治疗样本:ACRG/GSE62254, GSE57303, GSE84437, GSE15459, GSE26253, GSE29272, and TCGA-STAD。Affymetrix和Illumina生成的微阵列数据集的原始数据从Gene Expression Omnibus(https://www.ncbi.nlm.nih.gov/geo/)下载。在Affy软件包(23)中,使用用于背景调整的RMA算法处理来自Affymetrix的数据集的原始数据。使用RMA进行背景调整、分位数归一化以及使用中位数波兰算法对每个转录物的寡核苷酸进行最终总结。使用lumi软件包处理来自Illumina的原始数据。癌症基因组图集(TCGA)的数据从UCSC Xena浏览器(GDC hub)下载,详情见补充方法。对于TCGA数据集,RNA测序数据(FPKM值)被转换成每千碱基百万(TPM)值的转录物,这与微阵列产生的结果更相似,并且在样本之间更具可比性(24)。数据集选择的标准、每个数据集的平台和来源、样本数量和临床终点总结在补充方法和补充表S1中。使用R(3.4.0版)和R生物导体包分析数据。 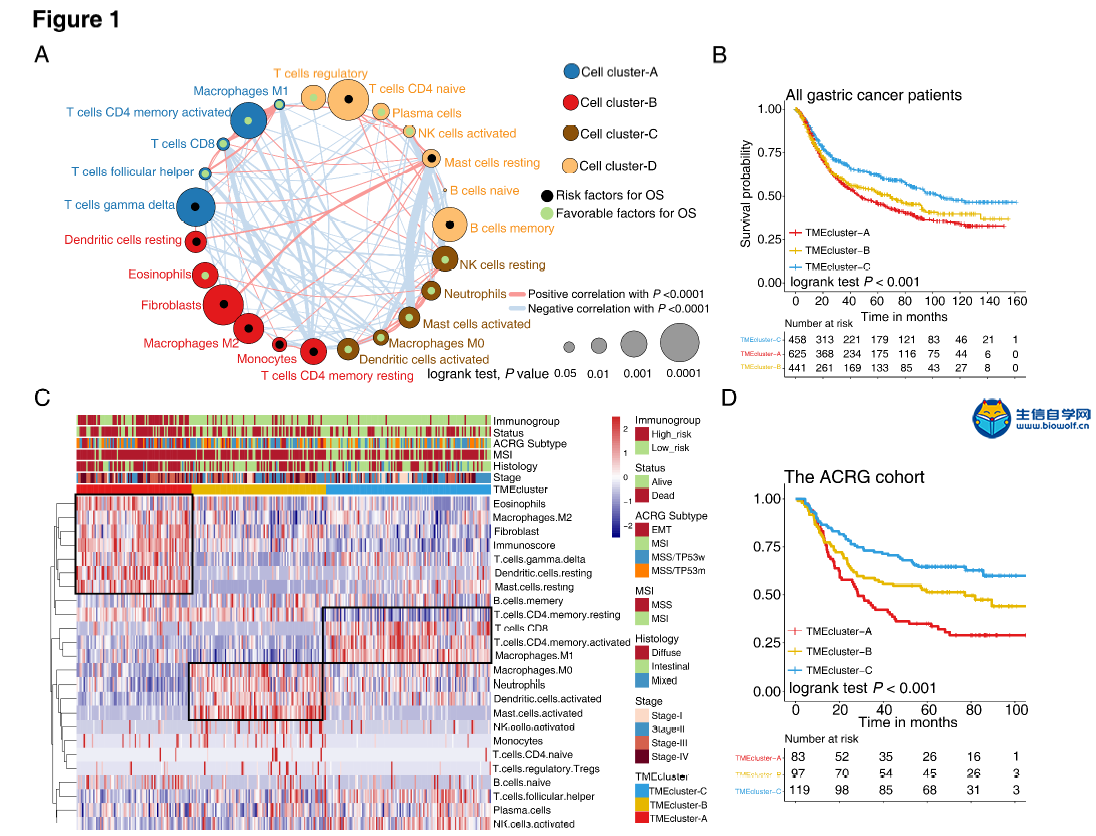 临床资料收集 从这些数据集中检索相应的临床数据,并在可用时手动组织。对于一些系列,未附在基因表达谱上的临床数据是通过以下三种方法之一获得的:i)直接从GEO数据集网站的相应项目页面下载,i i)从相关文献的补充材料下载,以及i i i)使用R中的GEOquery包。必要时联系了相应的作者以获取进一步的信息。更新的TCGA-STAD样本的临床数据和样本信息来自基因组数据共享(https://portal.gdc.cancer.gov/)使用R包TCGAbiolinks(25)。所有TCGA数据集的总体存活信息均来自最近发表的研究补充数据(26)。 TME中浸润细胞的推断 为了量化胃癌样本中免疫细胞的比例,我们使用CIBERSORT算法(16)和LM22基因标记,这使得对包括B细胞、T细胞、自然杀伤细胞、巨噬细胞、树突状细胞和髓系亚群在内的22种人类免疫细胞表型具有高度敏感和特异性的区分。CIBERSORT是一种反褶积算法,它使用一组参考基因表达值(一个带有547个基因的签名),被认为是每种细胞类型的最小表示,并基于这些值,使用支持向量回归从混合细胞类型的大体积肿瘤样本中推断出细胞类型比例。基因表达谱使用标准注释文件编制,数据上传到CIBERSORT网站(http://cibersort.stanford.edu/),算法使用lm22签名和1000个排列运行。通过应用微环境细胞群计数器方法估计基质细胞的比例,该方法允许根据转录组数据对异质组织中8个免疫细胞群和2个基质细胞群的绝对丰度进行稳健量化(17)。 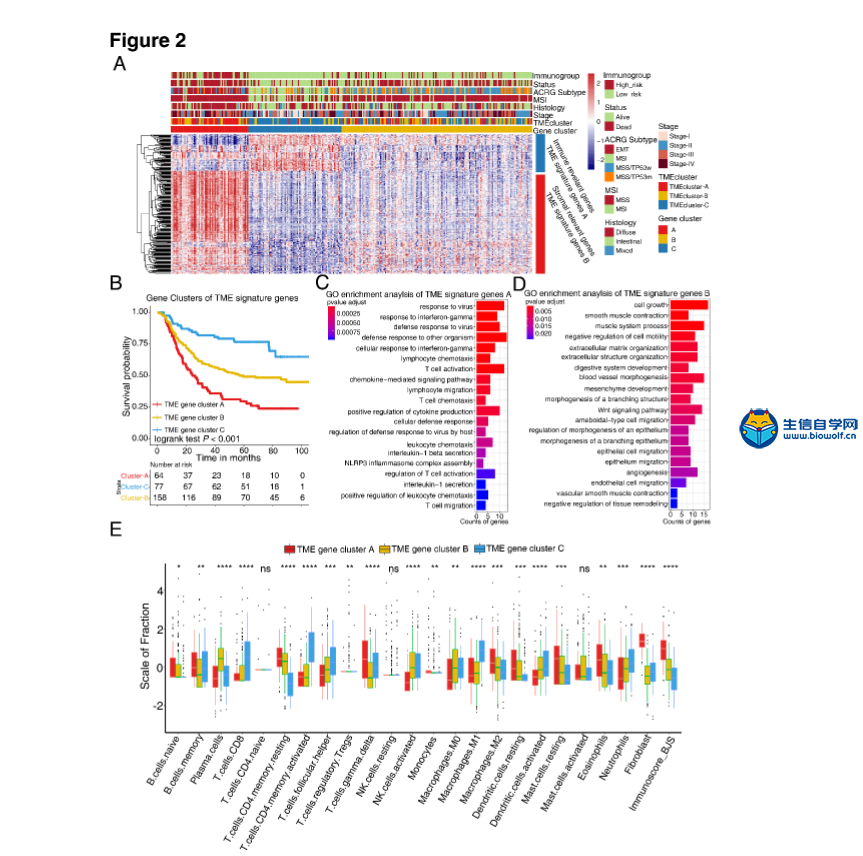 TME浸润细胞的共识聚类 利用层次聚集聚类(基于Euclidean distance and Ward's linkage)对具有不同TME细胞浸润模式的肿瘤进行分类。采用无监督聚类方法(k-均值)(27)进行数据集分析,确定TME模式,并对患者进行分类,以便进一步分析。应用一致性聚类算法确定元数据集和亚洲癌症研究组(ACRG)队列中的聚类数,以评估发现的聚类的稳定性。该程序使用ConsenseClusterPlus R包(28)执行,重复1000次以确保分类的稳定性。 与TME表型相关的差异表达基因(DEGS) 为了识别与TME细胞浸润模式相关的基因,我们将患者分为TMEcluster-A, TMEcluster-B, 和TMEcluster-C三组。使用limma(29)R包确定这三组患者中的DEGs,该方法使用适度的t检验来估计基因表达的变化。TME亚型间的DEG由limmaR包中实施的显著性标准(矫正后p值<0.05)确定。使用Benjamini Hochberg校正(30)计算多次试验的矫正后P值。 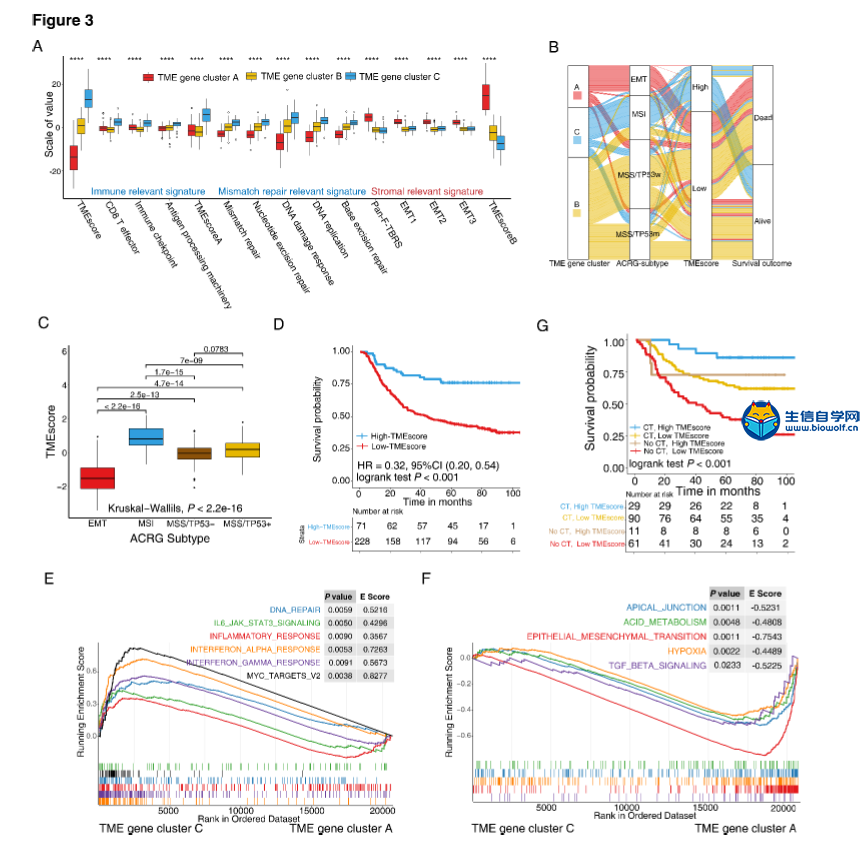 TME基因标记的降维与生成 TME基因的构建过程如下。首先,在ACRG队列的所有样本中,TMECluster-ABC中的每个DEG都被标准化。采用无监督聚类法(K-均值)(27)对DEGs进行分析,将患者分为三组进行进一步分析。然后,采用随机森林分类算法进行尺度约简,以减少噪声或冗余基因(31)。接下来,采用clusterprofiler R包(32)来注释基因模式。采用一致性聚类算法(28)定义基因聚类,并进行主成分分析(PCA)。提取主成分1作为基因标记分。在获得每个基因标记评分的预后值后,我们采用类似于GGI(33)的方法来定义每个患者的TMEscore: TMEscore = Σ PC1i –ΣPC1j 其中i是cox系数为正的簇的特征值,j是cox系数为负的基因的表达水平。补充方法中描述了详细的数据预处理步骤。 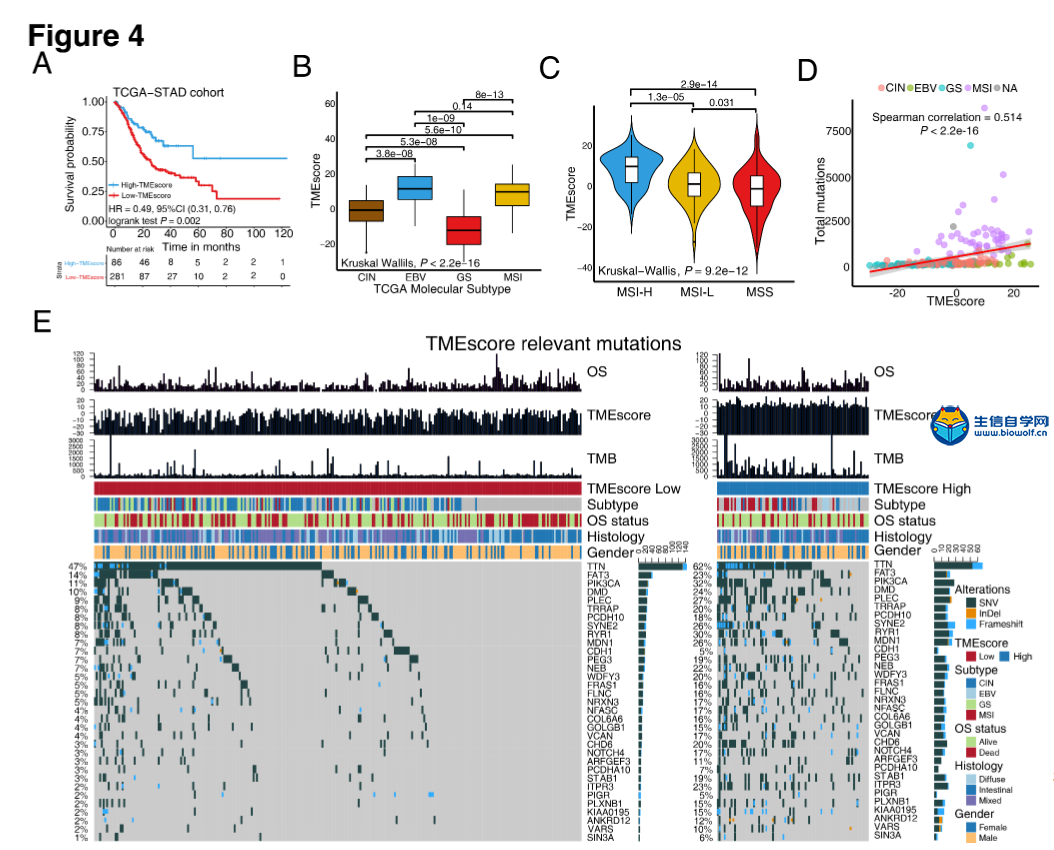 功能和途径富集分析 使用clusterprofiler R包(32)对TME标记基因进行基因注释富集分析。GO富集的确定严格限制在P<0.01,错误发现率(FDR)小于0.05。我们还通过对所有转录物的调整后表达数据进行基因集富集分析(GSEA)(34),确定了TME基因簇A和C对特定TME表型上调和下调的途径。基因集从Broad研究所的MSigDB数据库下载(34)。我们从策展基因集/典型途径收集中包括广泛的特征和特定的兴趣途径。富集P值基于10000个排列,随后使用Benjamini-Hochberg程序调整多次试验,以控制FDR(30)。 免疫检查点阻断的基因组和临床数据集 5份转移性尿路上皮癌(13)患者用抗程序性死亡配体1(pd-l1)试剂(阿托唑单抗)治疗的基因组和转录组学数据集,程序性死亡1(PD-1)阻滞剂(35)治疗晚期黑色素瘤患者,来自TCGA-SKCM队列的不同类型免疫治疗的晚期黑色素瘤患者(36),采用MAGE-A3抗原免疫治疗(37例)和抗CTLA4抗体(38例)小鼠模型治疗晚期黑色素瘤,然后分析了TME签名分数的预测值。补充方法中详细介绍了数据源和预处理方法。 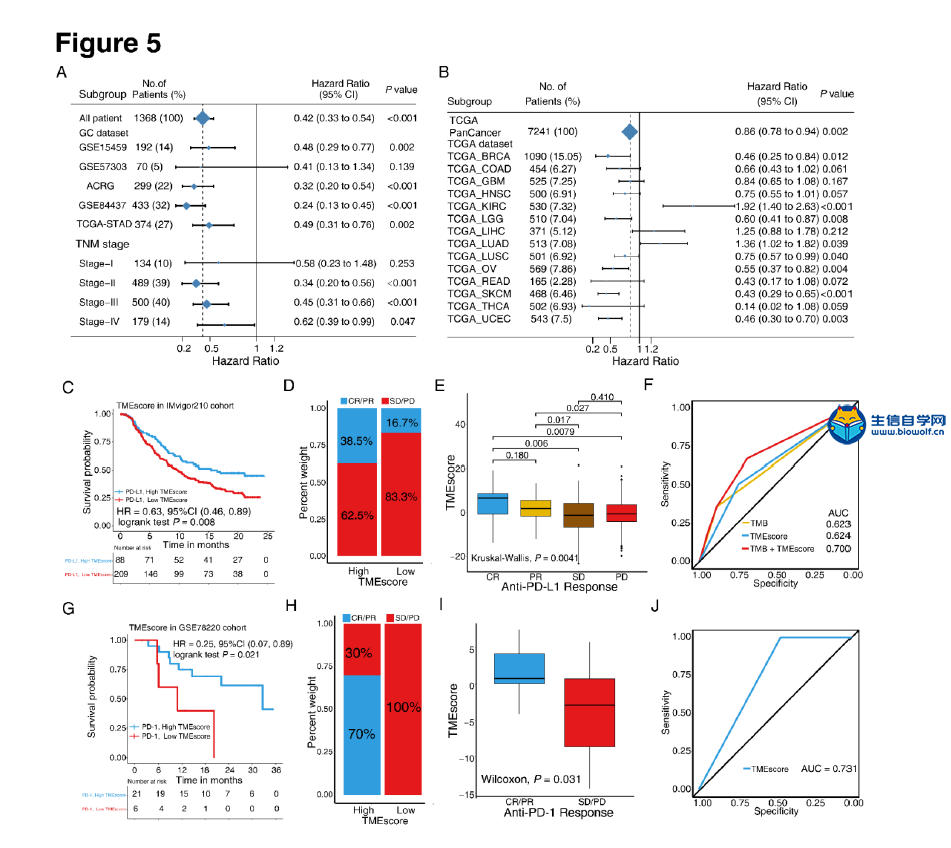 统计分析 采用Shapiro-Wilk正态检验(39)检验变量的正态性。对于两组的比较,用不配对的t检验估计正态分布变量的统计显著性,用Mann-Whitney U检验(也叫Wilcoxon秩和检验)分析非正态分布变量。对于两组以上的比较,分别采用Kruskal-Wallis检验和方差单向分析作为非参数和参数方法(40)。相关系数由Spearman和距离相关分析计算得出。采用双侧Fisher精确检验分析应急表,利用survminer软件包,根据患者总生存率与各独立数据集TMEscore之间的相关性,评估各数据集的截止值。使用MaxStat(41)R包对所有可能的切点进行迭代测试,以找到达到最大秩统计的切点,将TMEscore进行二值化,然后将患者分为低和高TMEscore亚型。为了识别差异基因分析中的重要基因,我们采用Benjamini-Hochberg方法将P值转换为FDRs(30)。采用Kaplan-Meier方法生成各数据集各亚组的生存曲线,采用对数秩(Mantel-Cox)检验确定差异的统计显著性。使用单变量Cox比例风险回归模型计算单变量分析的风险比。采用多变量Cox回归模型确定独立的预后因素。利用pROC R包(42)绘制和可视化接收机工作特性(ROC)曲线,计算曲线下面积(AUC)和置信区间,评价TMB、TMEscore及其组合的诊断准确性。为了比较AUC,采用了两条相关ROC曲线的似然比检验。所有统计分析均使用R(https://www.r-project.org/)或SPSS软件(版本25.0)进行,P值为双侧检验。小于0.05的P值被认为具有统计学意义。 对的,上面就是这篇高分文献的基本方法 有需要做生信报告合作的学员,请直接联系微信:18520221056  
责任编辑:伏泽 作者申明:本文版权属于生信自学网(微信号:18520221056)未经授权,一律禁止转载! |




